EMRinger: a side-chain-directed approach to study model-to-map agreement in cryo-EM
17 February 2015
For the past year, I have been working in the Fraser Lab on developing an analysis framework, which we call EMRinger, for doing model-to-map validation in the burgeoning field of near-atomic-resolution single particle electron cryomicroscopy (cryo-EM). We recently submitted our paper for review; at the same time, we preprinted it in bioRxiv (doi: 10.1101/014738) and open sourced the code. We hope that even as the article undergoes the peer review process, the tool will be available for scientists hoping to get an independent metric for progress in their refinement. We also hope people will be able to start including it as a “Table 1” metric now that the code and the manuscript are available. We have already been using EMRinger with our collaborators for a few months, and we are excited to see how it gets used now that it is out in the wild!
The manuscript is the best place to get scientific details about the work, but I am writing this post to talk informally about the method, as well as the process of developing it and the preprinting experience.
The background: ab initio modeling in cryo-EM
In the past two years, electron cryomicroscopy has been able to achieve resolutions at which atomic models can begin to be inferred without any sort of crystallographic reference structure, as a result of major developments both in camera technology and computational analysis. Electron microscopy holds tremendous promise for structural biology because of its ability to solve structures with minimal amounts of protein and without needing to find conditions for crystallization. Already, it has become one of the preferred methods for solving structures of large membrane proteins and biological complexes. However, the tools for validation of structures from cryo-EM are in their infancy relative to diffraction methods, and there are different requirements for validation compared to diffraction. Our tool takes advantage of the density around side chains, the highest resolution information available, to sensitively report on backbone positioning in the map.
EMRinger - using side chain information to sensitively report on backbone position.
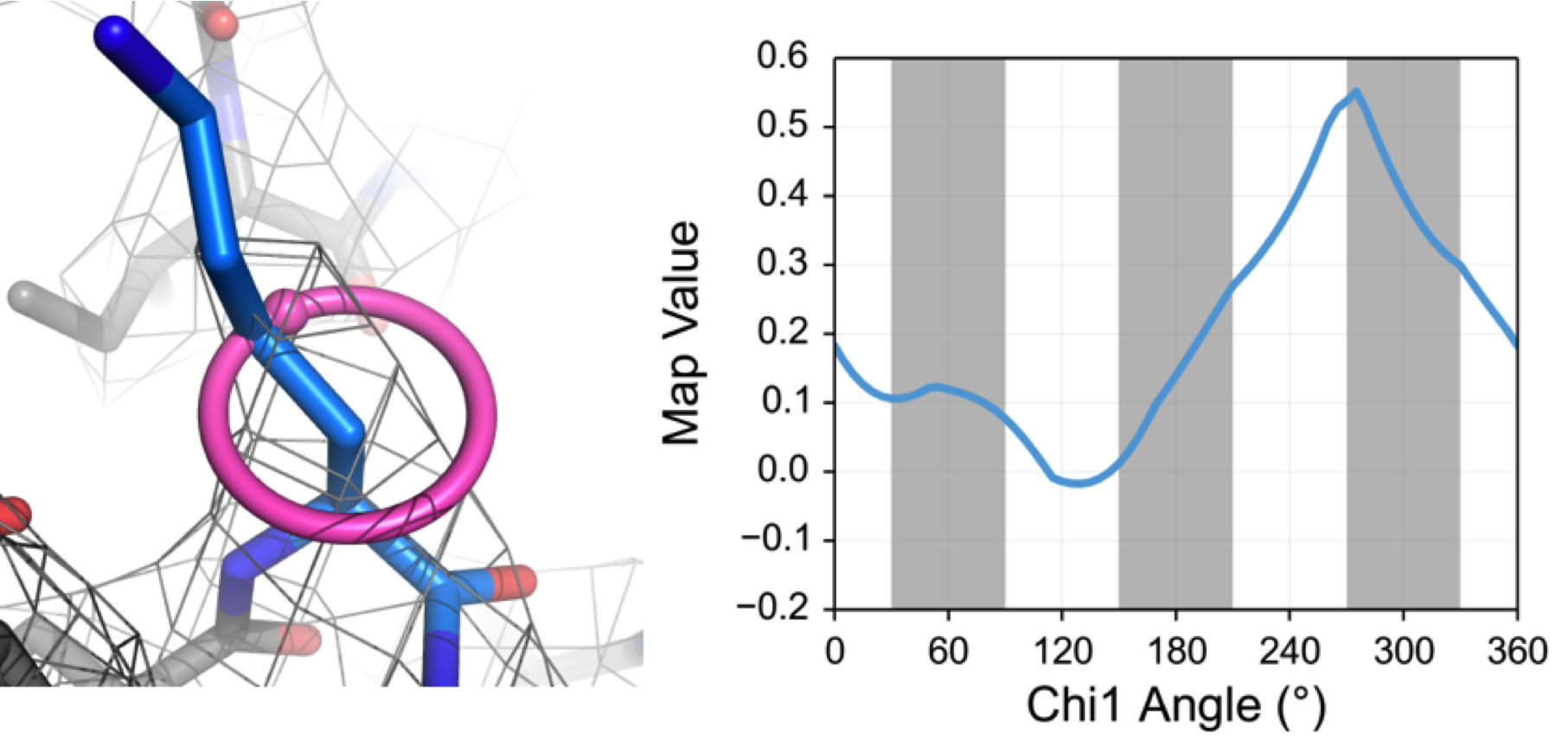 EMRinger is named after its progenitor, Ringer, from the Alber lab at Berkeley. Like Ringer, it rotates the Cγ atom around the χ1 angle of a side chain, interpolating the density value in the map as it rotates. The angle of peak density is interpreted as the correct position of the side chain in dihedral space. Based on knowledge from performing Ringer on structures in the PDB, as well as from basic steric considerations, we know that properly modeled side chains should have χ1 density peaks at or near rotameric positions (60º, 180º, and 300º). There are two major reasons that the peak may not be rotameric: first, if there is no signal above noise from the side chain, the peak will be chosen randomly from the noise and may be rotameric, and second, if the backbone and Cβ positions are incorrect, the measured angle relative to that backbone may be reported as nonrotameric when the Cγ samples its true position. We take advantage of this second expected behavior to quantify the quality of model-to-map fit of models from cryo-EM.
EMRinger is named after its progenitor, Ringer, from the Alber lab at Berkeley. Like Ringer, it rotates the Cγ atom around the χ1 angle of a side chain, interpolating the density value in the map as it rotates. The angle of peak density is interpreted as the correct position of the side chain in dihedral space. Based on knowledge from performing Ringer on structures in the PDB, as well as from basic steric considerations, we know that properly modeled side chains should have χ1 density peaks at or near rotameric positions (60º, 180º, and 300º). There are two major reasons that the peak may not be rotameric: first, if there is no signal above noise from the side chain, the peak will be chosen randomly from the noise and may be rotameric, and second, if the backbone and Cβ positions are incorrect, the measured angle relative to that backbone may be reported as nonrotameric when the Cγ samples its true position. We take advantage of this second expected behavior to quantify the quality of model-to-map fit of models from cryo-EM.
Generating the EMRinger score
In order to separate the effects of noise from the effects of mispositioned backbone atoms, we implement a signal cutoff threshold for peaks, with the assumption that any side chain with a peak map value below that threshold may be sampling noise and should not be used for analysis. For the remaining side chains, we use the fraction of side chains with rotameric peaks from EMRinger as a metric for the quality of model-to-map fit.
Choosing the right threshold is a challenging question; at too low of a cutoff, peaks are coming from noise as often as from real signal, and at too high of a cutoff, very few side chains are sampled and the results are not very representative of the model as a whole. In order to balance these two considerations, we use a Zscore metric based on the null hypothesis of a random binomial distribution with 39 out of 72 bins being called rotameric.

The Z-score is a good metric that is (in our experience) always able to find an ideal threshold to maximize the statistical significance of the result. However, it is biased towards models which are larger to begin with; a 60-mer would have a score more than 7 times larger than the equivalent monomer. To account for this, we rescale the Z-score to the EMRinger Score, which is comparable between sample sizes.

The maximum EMRinger score obtained across threshold is the one we use as our final metric, and our evidence (as we discuss in the manuscript) is that it is a robust but sensitive metric for model-to-map fit in very high resolution maps, above around 4 Å. The EMRinger scores of model-map pairs from the EMDB sets up a reasonable expectation for what scores should look like for well-refined structures at high resolution:
There is a strong correlation between resolution and EMRinger score. This is to be expected, given that EMRinger score reports on side chain density, which is only resolvable above about 4.5 Å. In general, for maps above around 3.5 Å resolution, the minimum score that should be expected is around 1, with a benchmark for a very good score lying around 2. Most structures which have been carefully refined, either in real or reciprocal space, score above 1.5, with some structures getting scores above 3. Just like with tools like molprobity, a bad score doesn’t necessarily mean an unusable structure, but it does warrant further consideration and refinement.
At resolutions down near 4 Å and below, things get a little bit trickier, and it is hard to make careful predictions about what the target EMRinger score should be. Sampling of the backbone by EMRinger becomes a problem at these resolutions, which can be partially resolved through B-factor sharpening. We are considering ways to modify the EMRinger procedure to make it more appropriate for use in these intermediate resolutions in the future, and I will hopefully write more about that soon!
Making EMRinger Scores Available
Since the score is straightforward to calculate and better scores are strong indicators of a better-fit model, we hope that this tool will be adopted as a standard “Table 1” metric for structures from cryo-EM, alongside gold-standard FSC resolution and Molprobity scores. To that end, we have made the tool available to colleagues as quickly as possible upon completing the manuscript: we have preprinted our work, and we have published and open sourced our code immediately.
Preprinting with bioRxiv
The first and most important thing we did to increase the availability of EMRinger was to preprint the paper on bioRxiv as soon as we submitted the manuscript for review. BioRxiv is a service maintained by Cold Spring Harbor which allows the public deposition of a manuscript before, or even entirely separately from, the peer review process. This allows us to publically share the work we have been doing without waiting for the peer review process, which has been known to take over a year before publication. Additionally, people unrelated to the official peer review process, but who have suggestions for strengthening the method, can provide commentary on bioRxiv that we can then take into account when revising our paper. The Fraser Lab has recently gotten excited about bioRxiv, and we plan on releasing many of our future papers this way. With many (most?) journals now allowing preprinting of manuscripts before peer review, there is little to lose in doing so. So far, we have primarily heard from colleagues directly about our manuscript, but we are excited to see what comments come out of the public forum available on bioRxiv.
We have been really happy with the reception of the paper on bioRxiv; we’ve had about 80 downloads of the paper in the two weeks since we submitted it. James and I have immediately become obsessed with the article metrics available on the site (particularly the altmetric). We are big twitter users and we take every chance we get to share our progress on EMRinger now that it is available in a public form. More importantly, we’ve been able to share the method directly with our colleagues, and if they want to use the EMRinger Score in their publication they will be able to cite the manuscript on bioRxiv ahead of formal publication.
Publishing code on Github
Along with making the manuscript available, we have released the code to run EMRinger on Github. We have published the main utilities to calculate EMRinger score, perform rolling-window analysis, and confine EMRinger analysis to specific residue groups, which are the main ways we have used EMRinger and expect it to be used in the near future. I am also in the process of cleaning up some of my “jiffy” scripts to generate the figures we used in the manuscript, so that the connection between the output of the EMRinger code and the figures we present is as clear as possible. We hope this will also allow anyone interested in extending the usage of EMRinger into new domains to visualize their results quickly.
I wrote the EMRinger scripts in python using the Phenix software package, which provides a whole host of incredibly nice utilities for working with structural biology data. Phenix has so far been used mostly for data from diffraction experiments, but there are an increasing number of tools available for single particle experiments, and I am hopeful that Phenix will be able to bring together experimentalists from both fields. This has the potential to develop standardization in the tools for modeling and analysis, as well as in the data formats between the two fields.
I wrote a detailed usage guide for generating EMRinger Scores in the Phenix_Scripts folder of the EMRinger github page, where the main utility scripts for EMRinger are housed. However, if you try it out, and encounter any problems that are not covered in the usage guide, or just want to discuss how best to interpret your results, I am happy to communicate by email. Please submit bug reports or feature requests on the github, and general comments on the biorxiv page!
Next steps: Phenix GUI and EMRinger Web Server
We are very happy with the Phenix python scripts on github, but in the long term we want to make our tool as universally accessible as possible, without any need to run scripts from the command line. We have been working hard on making EMRinger more accessible in two main ways. The first is putting the tool into the Phenix GUI: this effort is being driven primarily by our collaborator, Nathaniel Echols. Bringing EMRinger into the GUI will allow the rapid calculation of EMRinger scores, as well as making interactive visualization of the ringer scans possible. Since many other analysis tools, including molprobity, are accessible within Phenix, we hope that this inclusion will allow electron microscopists to do every level of model-based validation within a single software package, without needing to resort to individual scripts.
The second way we are trying to expand the availability of EMRinger is by making a webserver to calculate EMRinger scores on demand. I have been working on this as a side project for a few months, and actually got a proof of principle iteration done as part of a minicourse at UCSF with Kevin Hartman and Andrew Van Benschoten. Users will be able to upload their model and map and the webserver will return a series of visualizations as well as the final EMRinger score. I hope that this serves as the most accessible introduction possible to using EMRinger, as the only software needed will be a modern web browser. I’ll be open sourcing it as soon as I have a working prototype (soon, I hope!), and I’ll probably write another post then about the process of making a scientific webserver. There are unique scalability challenges to operating a webserver that can handle map data (which is frequently hundreds of megabytes in file size), and I hope that the solutions that we come up with will allow for a robust system for electron microscopy analysis on the web.
Citation: Barad BA, Echols N, Wang RYR, Cheng Y, Dimaio F, Adams PD, Fraser JS. (2015). Side-chain-directed model and map validation for 3D Electron Cryomicroscopy. biorXiv doi: 10.1101/014738
comments powered by Disqus